Francis Collins, and many others, believe that the concept of junk DNA is outmoded because recent discoveries have shown that most of the human genome is devoted to regulation. This is part of a clash of worldviews where one side sees the genome as analogous to a finely tuned Swiss watch with no room for junk and the other sees the genome as a sloppy entity that's just good enough to survive. (pp. 264-266)What is regulation?
Regulation refers to gene expression that can be modified according to environmental conditions. (pp. 266-267) [Protein concentrations in E. coli are mostly controlled at the level of transcription initiation]Stochastic gene expression
The rate of transcription of a gene can vary from cell to cell due to the stochastic nature of transcription factor binding and the initiation of RNA synthesis. This is not regulation. (pp. 267-268)What do we know about regulatory sequences?
Most transcription factors bind within a few hundred base pairs of the promoter. With a few exceptions, regulatory sequences are found in close proximity to the 5′ end of the gene. (pp. 268-270) [Are multiple transcription start sites functional or mistakes?] [How many enhancers in the human genome?] [Are most transcription factor binding sites functional?] [The Encyclopedia of Evolutionary Biology revisits junk DNA]
Box: The making of a queen by regulating gene expression (p. 270)
Regulation and evolutionOne way of resolving the Deflated Ego Problem is to speculate that humans have a much more sophisticated regulatory network than other "lower" species. This is consistent with evo-devo, which postulates that the differences between species is due more to differences in regulating gene expression that differences in the number of genes. However, most of the results from evo-devo suggest that the differences in regulation are due to very small changes in the binding of existing transcription factors and not huge changes in genome organization. (pp. 271-273)
Box: Can complex regulation evolve by acccident? (pp. 273-274)
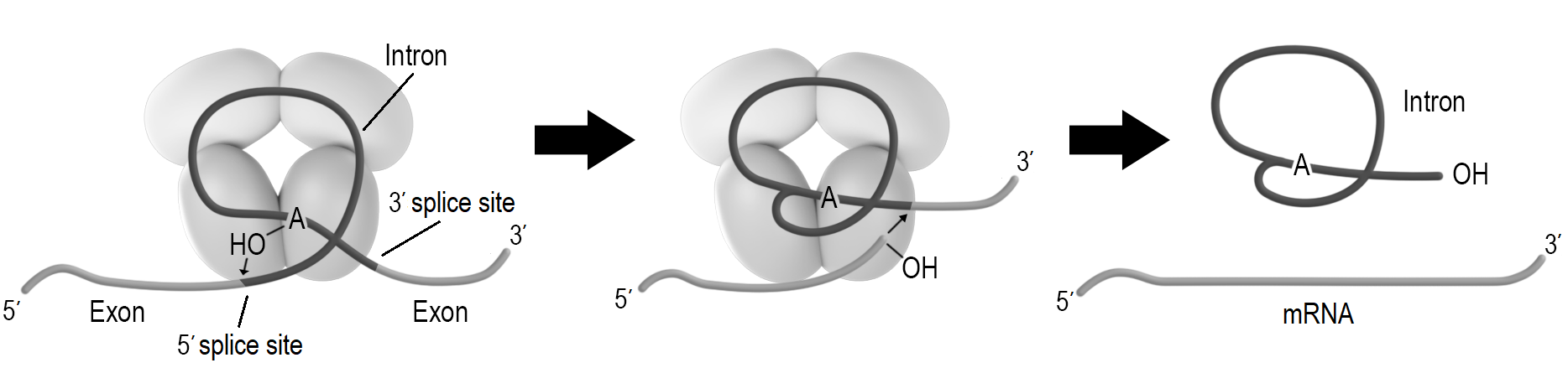
Regulating gene expression by rearranging the genomeThere are some well-studied examples of regulation that are connected to rearranging the genome by recombination. (pp. 275-277)Open and closed domains
DNA is more accessible to transcription factors when nucleosomes are loosely organized in an open domain. Gene expression is repressed when the gene is embedded in a highly structured closed domain (heterochromatin). The transition from closed to open domains is coupled to demthylation of DNA and modification of histones. The DNase I sensitivity of DNA in an open domain is correlated with transcription activity. The spontaneous "breathing" of heterochromatic regions allows transcription actors to bind. (pp. 277-279) [Epigenetic markers in the last 8% of the human genome sequence] [Chromatin organization at promoters in yeast cells]
Box: X-chromosome inactivation (pp. 279-280) [Escape from X chromosome inactivation
The recruitment model of gene expressionThe recruitment model of gene expression says that the binding of transcription factors triggers the demethylation of DNA and the modifiction of histone proteins to maintain an open domain. The histone code model is often connected to the belief that DNA demethylation and histone modification are the key events in regulation and not epiphenomena. This view is usually associated with a strong belief in the importance of epigenetics. (pp. 281-282ENCODE promotes regulation
ENCODE researchers postulate that at least 20% of the genome is required for regulation and there are dozens of transcription factor binding sites for each gene. According to this view, this sophisticaed regulation explains why complex humans can exist with the same number of genes as most other species. (pp. 283-285) [ENCODE's false claims about the number of regulatory sites per gene]

There are very few known examples of human genes with the complicated regulatory mechanisms promoted by the ENCODE leaders. The few published genomics tests of the hypothesis do not support it. What's missing is the random genome project in order to emphasize the importance of a negative control. (pp. 285-287) [How much of the human genome is devoted to regulation?]
Box: A thought experiment (pp. 287-288)
3D chromosomesOne possibility is that a lot of extra DNA is required in humans in order to organize genes into large functional loops of chromatin. This idea has been promoted by several scientists, including Emile Zuckerkandl. (pp. 288-291)What the heck is epigenetics?
The most useful definition of epigenetics is the Holliday definition that restricts the term to changes that could be inherited by daughter cells following cell division. Proponents of epigenetics claim that chromatin markers can be passed from generation to generation in humans and they determine whether a gene will be expressed or silenced. There is no known mechanism for passing such markers from somatic cells to the germ line. (pp. 292-293) [What the heck is epigenetics?] [Nessa Carey talks about epigenetics] [What do believers in epigenetics think about junk DNA?]

The restriction/modification system in bacteria is a good example of how methylation signals can be passed to daughter cells following cell division but it does not explain epigenetics. There is a lot of hype associated with epigenetics and much of it is unjustified.(pp. 293-296)
Notes for Chapter 10 (pp. 331-333)